
发布时间: 2024-08-14 09:22:22
研究背景
虽然GERD患者肠道菌群研究取得了一些进展,但以往的研究大多集中于食管及胃内的微生物。研究表明,正常人食管菌群主要由厚壁菌门中的革兰氏阳性菌链球菌组成,但研究发现GERD患者食管内以拟杆菌门、变形菌门和梭杆菌门中的革兰氏阴性厌氧菌及微量需氧菌为主。此外,慢性炎症已被证明在多种胃肠道疾病(如BE和食管癌)的发展中发挥作用,而慢性炎症引起的肠道菌群改变则进一步加速疾病的发生发展。革兰氏阴性菌外膜的重要组成部分脂多糖(LPS)可能通过诱导NF-κB的表达来促进组织炎症。在动物模型中,高脂饮食通过调节肠道菌群上调IL-8/CXCL1炎症信号通路促进BE进展为食管癌,证实了肠道菌群在疾病进展中的作用。 GERD 患者肠道菌群的组成和功能仍不清楚,研究表明肠道菌群通过将宿主营养物质转化为代谢物在维持胃肠道稳态中发挥重要作用。食管的正常功能通过能量代谢、黏膜屏障修复和免疫调节等多种机制维持。因此,肠道菌群失衡导致代谢物平衡被破坏可能促进 GERD 的发生。
研究思路
连续入组30例GERD患儿和30例健康对照(HC),收集受试者的人口统计学和临床特征。首先采用16S rRNA测序评估GERD组和HC组患儿肠道菌群的差异,选取10例GERD患儿和10例HC组患儿进行宏基因组学分析。采用液相色谱/质谱法(LC/MS)进行非靶向代谢组学分析,并将宏基因组学和代谢组学数据结合起来分析。
主要结果
基于 16S rRNA 测序的 GERD 儿童肠道菌群组成变化
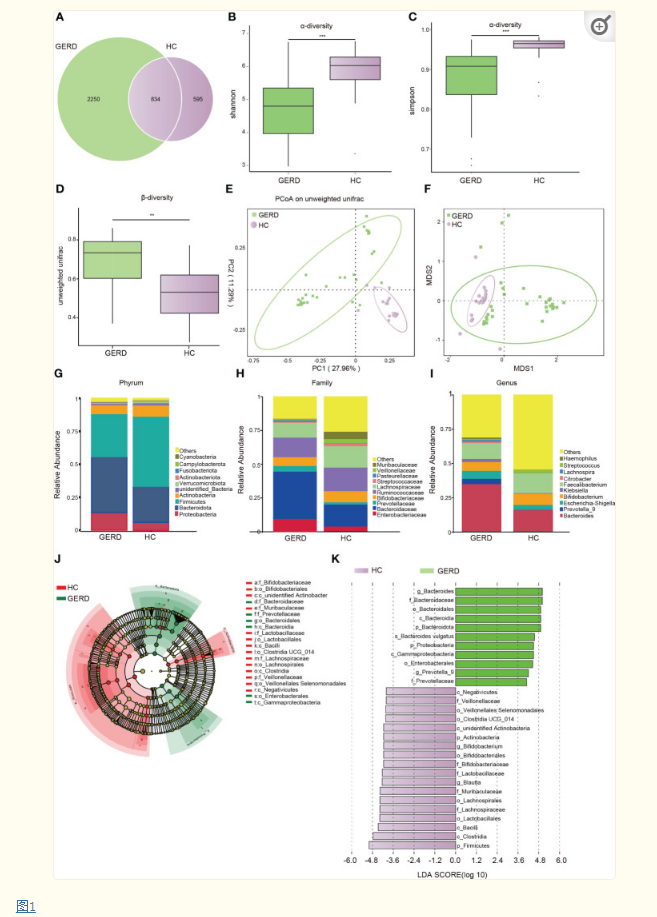
作者使用 16S rDNA 测序检测 GERD 儿童和 HC 儿童的粪便微生物。α多样性结果显示,GERD患儿的Shannon和Simpson指数显著低于HC组(图 1B、C),ANOSIM分析表明,两组微生物群落结构分布存在显著差异(图 1D),PCoA和NMDS显示两组肠道微生物组成存在差异(图 1E、F)。门水平上,GERD组变形菌门和拟杆菌门相对丰度显著高于HC组,而厚壁菌门和放线菌门相对丰度显著低于HC组(图 1G);在科水平上,GERD患儿肠杆菌科、拟杆菌科和普雷沃氏菌科相对丰度较高,双歧杆菌科、瘤胃球菌科和毛螺菌科相对丰度较低(图 1H);属水平上, GERD组患儿拟杆菌属、普氏菌属-9型、大肠杆菌-志贺菌属、克雷伯菌属和嗜血杆菌属的相对丰度均显著高于HC组患儿,而双歧杆菌属、粪杆菌属和链球菌属的相对丰度在GERD组低于HC组(图 1I)。LEfSe分析显示, GERD组中Prevotella-9和Bacteroides丰度最高,而HC组中Bifidobacterium和Blautia丰度最高(图 1J、K)。
宏基因组测序显示 GERD 儿童与 HC 儿童的肠道菌群组成存在明显差异
为了从物种水平上了解GERD患儿肠道菌群的变化,作者还进行了宏基因组测序。 PCoA 结果显示,GERD 组和 HC 组在物种水平上的微生物组成存在明显分离(图 2B),ANOSIM证实了两组肠道菌群组成在物种水平上存在显著差异(图 2C)。Bacteroides stercoris、Bacteroides vulgatus、Alistipes putredinis、Bacteroides dorei、Bacteroides fragilis的丰度显著增加,而Fusicatenibacter saccharivorans、Blautia wexlerae、 Blautia obeum、Anaerostipes hadrus、Eubacterium hallii的丰度显著降低(图 2E、F)。
GERD患儿肠道菌群的功能变化
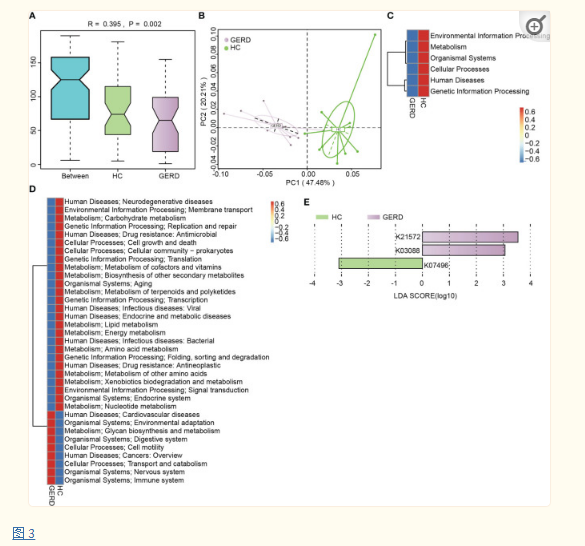
为明确GERD患儿肠道菌群的功能特征,本研究利用KEGG数据库进一步注释了肠道菌群宏基因组学数据,ANOSIM分析显示,在KEGG直系亲属(KO)水平上,GERD组与HC组肠道菌群存在显著差异(图 3A)。PCoA结果证实GERD组与HC组之间基于KEGG模块的微生物功能分布存在显著差异(图 3B),GERD组能量、氨基酸、维生素、碳水化合物、脂质等代谢途径活性低于HC组,而糖类合成及代谢途径活性高于HC组(图 3C-E)。
GERD患儿的异常代谢模式
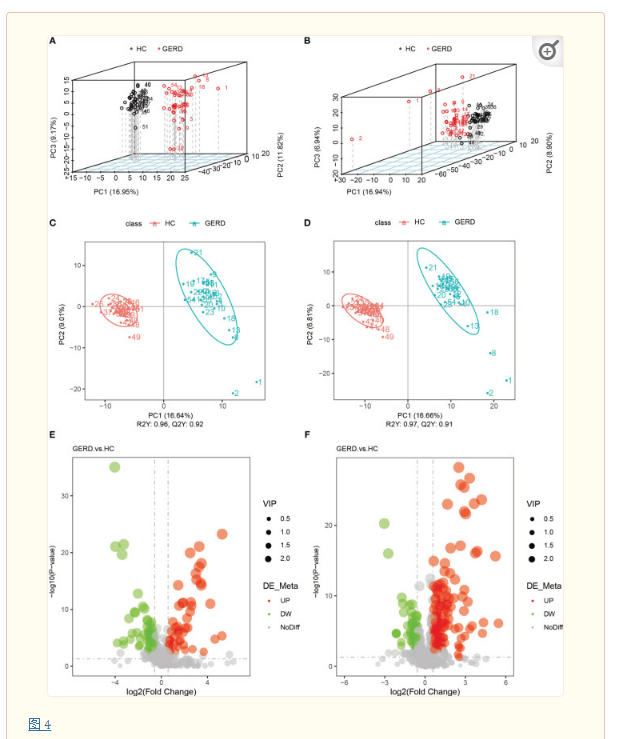
本研究还利用LC/MS技术对两组患儿血清样本进行代谢组学分析。PLS-DA结果显示GERD组与HC组血清样本代谢特征存在明显差异(图 4A–D)。进一步用火山图分析两组间的差异代谢物,阈值设定为VIP>1.0、FC>1.5或FC<0.667,P值<0.05。
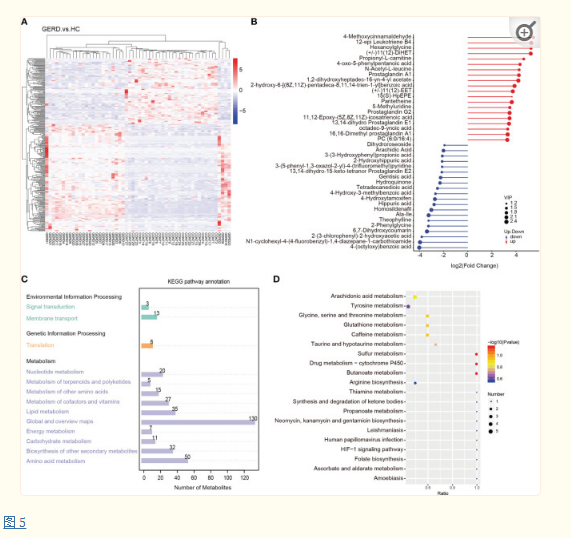
进一步对差异代谢物进行KEGG注释,发现富集排名前三的通路为全局及概览图、氨基酸代谢、脂质代谢(图 5C)。GERD患儿与HC患儿代谢物差异影响的代谢通路主要包括花生四烯酸(AA)代谢、酪氨酸代谢、硫代谢、甘氨酸、丝氨酸和苏氨酸代谢、谷胱甘肽代谢、咖啡因代谢(图 5D)。
肠道微生物与代谢物水平的相关性分析
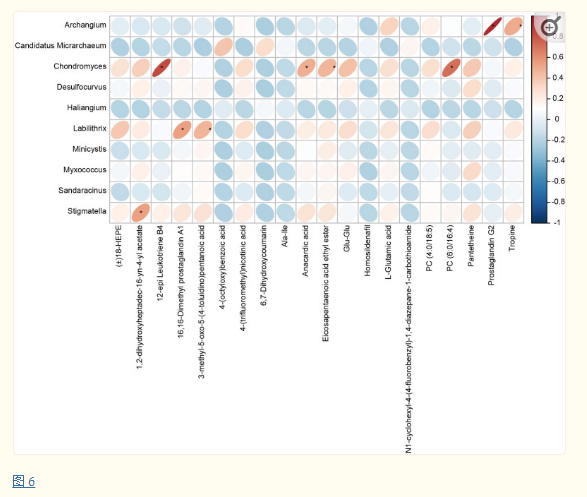
采用Pearson相关性分析方法分析GERD患儿肠道差异微生物与血清代谢物的关联性,结果显示GERD患儿血清代谢物的变化与肠道菌群中多种细菌水平的变化呈显著相关性,其中软骨霉菌(Chondromyces)、Labilithrix和Stigmatella的水平与多种代谢物水平呈显著正相关(图 6),表明肠道菌群和代谢物之间存在显著的相互作用,可能影响GERD的发生。
文章小结
本研究不仅揭示了GERD患儿肠道菌群失衡的现象,还揭示了GERD患儿肠道菌群中特定核心菌群的失衡及其功能变化,并进一步分析了肠道菌群与代谢物变化之间的关系,这些结果不仅为GERD相关肠道菌群失衡的假说提供了直接证据,也可能为进一步研究GERD的发病机制提供有力证据。
上一篇:肿瘤领域使用最广泛的公共数据库:监测、流行病学和最终结果数据库(SEER数据库)

